H3K27me3 spreading organizes canonical PRC1 chromatin architecture to regulate developmental programs
个人文献精读笔记,用于梳理论文证据链、图像逻辑、方法设计和可迁移研究思路。
Krug et al.; corresponding author: Nada Jabado
Nature Genetics, 2026
Morgen · tomorgene.com
个人文献精读 / Research Note
2026-06-30
2026-06-30
版本说明:本页为 tomorgene.com 公开导读版,保留机制梳理与读图逻辑,并含少量低分辨率示意图。含全部原图的完整精读稿仅供课题组成员等授权读者通过受控入口使用,不公开展示。如需访问权限,请通过 About 页联系方式联系。
版权与免责声明见页面底部。本页为第三方个人学习笔记,非 Nature / Springer Nature 官方内容。
一句话总结
这篇文章提出:不是单纯“H3K27me3 多或少”决定 Polycomb 靶基因沉默,而是 H3K27me3 在基因组上的“集中还是铺开”决定 cPRC1 的局部浓度,进而决定 Polycomb 远距离 3D loop、发育基因沉默和 H3K27M 胶质瘤的分化阻滞。
证据链阅读顺序
- 背景:PRC2 写入 H3K27me3,cPRC1 通过 CBX/PHC 读取并聚集,先建立 Polycomb 读写分工。(导读)
- 现象:H3K27M / EZHIP / hiPS 等 confined 状态下,H3K27me3-marked CGI 之间 3D interaction 增强。(Fig.1)
- 机制:confined 使有限 cPRC1 集中于少数位点,局部浓度升高,而非总量简单增加。(Fig.2-3)
- 因果:促进 H3K27me3 spreading 或打散 cPRC1,均可削弱 loop。(Fig.4)
- 功能:cPRC1 loop 压制发育分化程序并维持 H3K27M 肿瘤;CBX2/PHC2 扰动可部分逆转。(Fig.5-7)
名词解释
核心词支撑 Fig.1-7 主线;扩展词用于方法细节、疾病背景与 Extended Data。
核心概念 读懂主图必备
扩展概念 遇读再查
科学问题与总体模型
论文想解决什么
已知 Polycomb target 之间可以形成长距离 chromatin contacts,也已知 H3K27me3 能招募 cPRC1。但关键问题是:这些 contact 是怎样被调控的?它们是否只是表观相关,还是能真实影响发育基因沉默和疾病表型?
本文的巧妙切入点
作者没有只比较 H3K27me3 总量,而是抓住“分布形态”:H3K27M/EZHIP 肿瘤和 hiPS cell 代表 H3K27me3 confined;H3K27M-KO、oxygenation、NPC differentiation、NSD1-KO、EZH2-Y641N 等代表 H3K27me3 spreading 或 reduced confinement。
Fig.1 - H3K27me3 confined 状态对应更强 Polycomb 3D interactions
问题H3K27me3 restricted/confinement 是否和 Polycomb 靶位点之间的远距离 contact 有关?
读图要点:Fig.1a-b:BT245 中 PRDM13-SIM1 locus 的 Hi-C + H3K27me3 tracks + 3D DNA FISH。H3K27M 中两位点更接近;KO 后 H3K27me3 铺开,contact 变弱。
怎么看
- 上方 Hi-C 矩阵绿色圈代表目标 contact;H3K27M 中更强。
- 下方 ChIP-seq tracks 显示:H3K27M 是窄峰,KO 后 H3K27me3 domain 变宽。
- FISH 是独立验证:红绿 probe 距离在 H3K27M 中更短。
结论:H3K27M 并非简单造成全局染色质架构重排,而是在 H3K27me3-marked Polycomb loci 上增强特定长距离 contact。
读图要点:Fig.1c:aggregate peak / pile-up 思路。先定义 H3K27me3-enriched CGIs,再叠加位点对 Hi-C contact。
读图要点:Fig.1d:三个模型都展示 confined -> spread 的转换:H3K27M removal、EZHIP oxygenation、iPS-to-NPC。
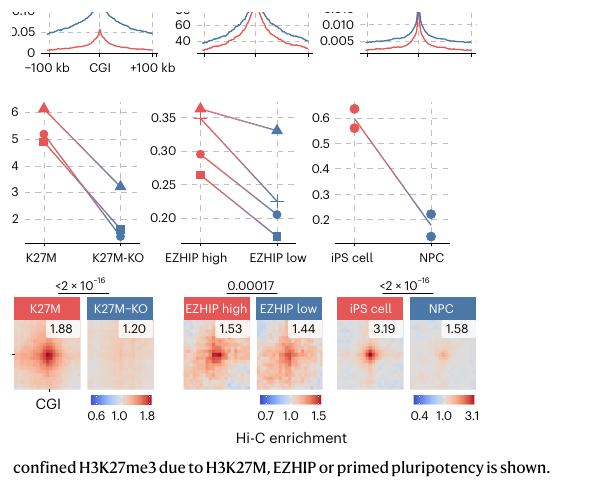
Fig.1e-g:confinement score 下降时,H3K27me3 CGI 之间的 Hi-C pile-up 中心信号同步下降。
Fig.2 - confined H3K27me3 会浓缩 cPRC1,而不是简单改变 PRC1 总量
机制如果 3D contact 更强,候选执行者是谁?作者把注意力放到 cPRC1 reader complex。
读图要点:Fig.2a-b:PRC1 组成示意 + chromatin fraction MS。H3K27M 降低 H3K27me3 总量,但 cPRC1 组分的染色质结合总池并没有剧烈下降。
读图要点:Fig.2c:CBX2/4/8 和 PHC2 高度重叠,定义 cPRC1 sites;RING1B 更广,提示还有 vPRC1 或其他 PRC1 活性。
读图要点:Fig.2d-e:CGI 层面,H3K27me3 retention 与 RING1B/CBX2 enrichment 相关;H2AK119ub 相关性弱。
读图要点:Fig.2f:代表 locus 显示 K27M 中 cPRC1 signal 更集中;KO 后 H3K27me3 broad spreading,cPRC1 被稀释到更宽区域。
关键理解
作者的“浓缩”不是说细胞里 cPRC1 总量变多,而是说有限的 cPRC1 在基因组上的分布更集中。这个概念很重要:同样数量的 reader,如果 landing pads 从少数窄峰扩展到大片 domain,局部浓度就会下降。
Fig.3 - cPRC1-rich sites 是 H3K27M 病理 Polycomb loop 的核心节点
分类作者把 CGIs/promoters 按 chromatin features 分成 active、cPRC1、PRC2-only、other,问哪一类位点真正贡献 3D loop。
读图要点:Fig.3a-b:UMAP/HDBSCAN 把 chromatin states 分类。cPRC1 cluster 同时带 H3K27me3、RING1B、CBX/PHC 等 Polycomb 特征,表达最低。
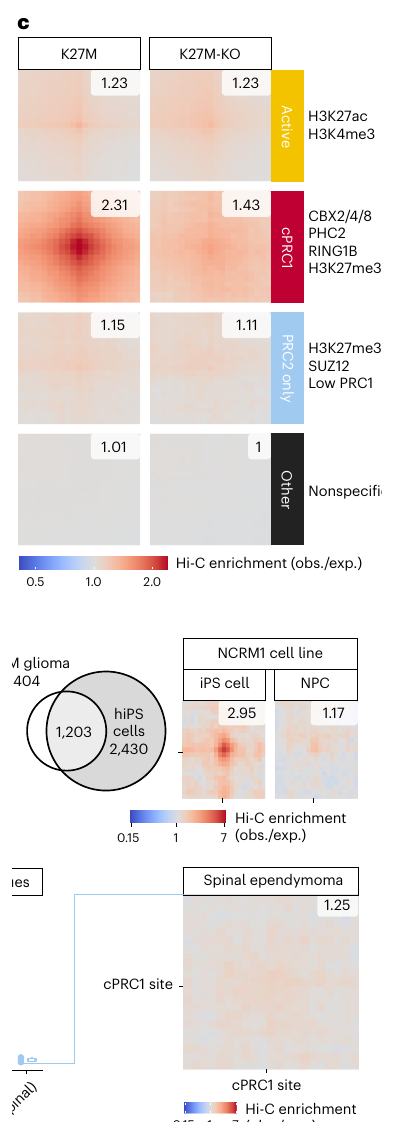
Fig.3c-f:只有 cPRC1 cluster 在 H3K27M 中显示显著 loop;该模式在 patient tissue、PFA-EPN 和 hiPS cell 中也强。
读图要点:Fig.3d-e:三个 H3K27M cell line 共有 1,607 个 consensus cPRC1 sites,并与 hiPS 的 cPRC1 target 有重叠。
读图要点:Fig.3f:原发组织中 H3K27M pHGG 和 PFA-EPN 的 cPRC1 site interactions 最强,说明不是细胞系假象。
逻辑推进:Fig.1 说 H3K27me3-CGI contact 强;Fig.2 说 cPRC1 在这些点被浓缩;Fig.3 进一步限定:真正形成强 loop 的,是 cPRC1-rich 的 Polycomb target subset。
Fig.4 - 反向扰动:让 H3K27me3 spread,会稀释 cPRC1 并削弱 loop
因果如果 confined H3K27me3 促进 loop,那么人为增加 spreading 应该削弱 loop。作者在 mES cells 里用 Nsd1-KO 和 EZH2-Y641N 做反向验证。
读图要点:Fig.4a:Nsd1-KO 后 H3K36me2 丢失,H3K27me3 spread,CBX2 signal 从 focal peak 变得更 diffuse,Nkx2-1/Foxa1 contact 变弱。
读图要点:Fig.4b-d:aggregate profile 与 chromatin state 分类显示,Nsd1-KO 主要削弱 cPRC1 cluster 的 loop。
读图要点:Fig.4e-g:EZH2-Y641N 作为另一种 H3K27me3 spreading 模型,同样降低 cPRC1 site contact frequency。
Fig.5 - cPRC1 loop 靶基因是被 H3K27M 压住的发育分化程序
功能前面证明了结构,接下来问:这些 loops 压住了什么基因?对肿瘤细胞状态有什么影响?
读图要点:Fig.5a-c:cPRC1 targets 可再分成 looping 与 other;looping genes 在 H3K27M-KO 后 nascent transcription 上升。
读图要点:Fig.5d-e:分化培养条件下,H3K27M-KO 对 cPRC1 loop genes 的上调更明显;体内 xenograft 中 KO 失去/延迟肿瘤形成。
读图要点:Fig.5f-g:scRNA-seq 显示 H3K27M 肿瘤更偏 progenitor-like,KO 后更偏成熟 glial states;cPRC1 loop gene expression 解除抑制。
读图要点:Fig.5h-j:在人类 bulk/scRNA patient data 中,H3K27M tumors 的 cPRC1 looping gene set 更低,支持临床相关性。
阅读要点:作者不是只看一个 marker,而是把 chromatin-defined cPRC1 loop gene set 投射到 bulk RNA、PRO-seq、xenograft scRNA 和 patient scRNA 多层数据中,形成结构到功能的闭环。
Fig.6 - 直接扰动 cPRC1 recruitment 或 aggregation,会削弱 loop 并促进分化
干预如果 cPRC1 是 loop 的执行者,那么阻断 CBX-H3K27me3 结合、敲掉 CBX2、或破坏 PHC2 oligomerization 都应该影响结构和功能。
读图要点:Fig.6a-b:CBX-AM、CBX2-KO、PHC2-L307R 都降低 cPRC1 loop pile-up,并提高 cPRC1 loop gene set activity。
读图要点:Fig.6c:SOX10 免疫荧光增强,说明这些扰动促进 oligodendrocyte precursor-like differentiation。
三个干预层次
- CBX-AM:药物阻断 CBX chromodomain 与 H3K27me3 的相互作用。
- CBX2-KO:去掉一个关键 cPRC1 reader/phase separation driver。
- PHC2-L307R:保留部分 chromatin occupancy,但破坏 SAM domain oligomerization,从而把“结合”和“聚集”拆开。
Fig.7 - CBX2/cPRC1 功能是 H3K27M 肿瘤维持的必要条件
读图要点:Fig.7a-c:CBX2-KO 或 PHC2-L307R 降低克隆形成;CBX2-KO xenograft 生存显著延长,组织中只有稀疏 engraftment。
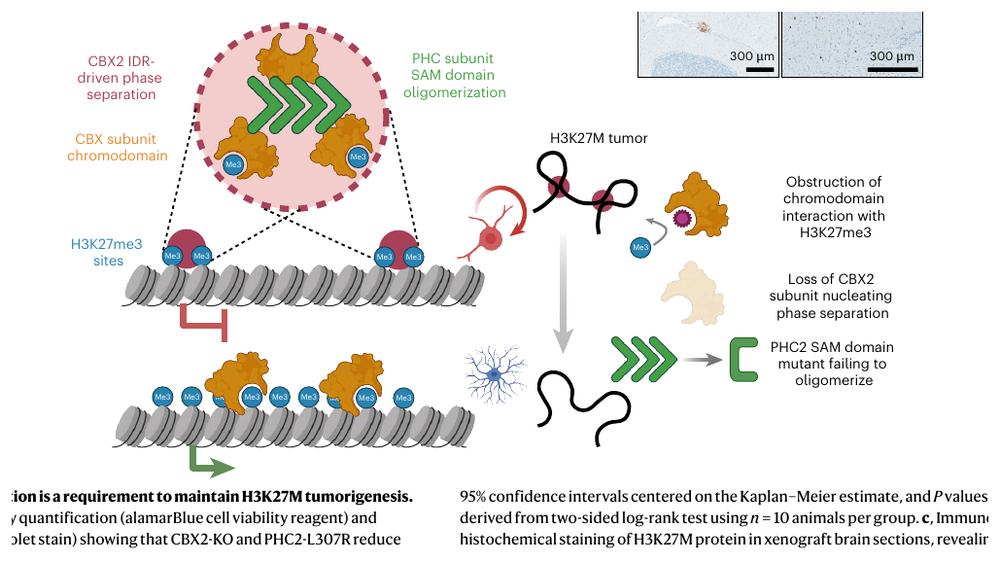
Fig.7d:模型图。H3K27me3 confined sites 上 cPRC1 foci 形成,三类 perturbation 都可打破 repressive loop architecture。
Extended Data 精读:哪些是必须讲的支撑证据
以下为支撑主图的补充证据;公开版从略,详见原文 Extended Data。
| 优先级 | 图 | 一句话 | 阅读取舍 |
|---|---|---|---|
| 必读 | ED1A-E | 排除 compartment、TAD、CTCF loop 等全局结构解释。 | 支撑“变化集中在 Polycomb/cPRC1 sites”。 |
| 必读 | ED6 | CBX-AM、CBX2/CBX4-KO、PHC2-L307R 的 controls。 | 判断 Fig.6 扰动是否可信。 |
| 重点 | ED1F-I | confinement score 技术验证与发育趋势。 | 理解 Fig.1 量化指标时读。 |
| 选读 | ED2 | PRC 总量和占位变化不能简单解释主效应。 | 用于回应“是不是总量变化”的质疑。 |
| 选读 | ED4 | 多 stem/cancer 模型支持 confinement 原则。 | 讨论机制泛化时读。 |
| 选读 | ED7 | CBX2 而非 CBX4 的体内特异性。 | 讨论 CBX paralog 差异时读。 |
读图要点:ED1A-E:H3K27M 并未在 compartment/TAD/CTCF loop 等全局层面形成清晰独立类别。支持“变化集中在 Polycomb/cPRC1 sites”。
读图要点:ED1F-I:confinement score 的技术验证,并显示小鼠脑发育过程中 H3K27me3 confinement 随时间下降。
读图要点:ED2A-D:PRC1/2 总 abundance 与 expression 大体不解释主效应;CBX2/RING1B occupancy 在 H3K27M 中更 confined。
读图要点:ED2E-H:独立 cell lines 中重复 H3K27me3-cPRC1 相关性;H2AK119ub 并不跟随同样模式。
读图要点:ED3:cPRC1 cluster expression 低;consensus cPRC1 targets 富集于发育和神经分化通路。
读图要点:ED4:NSD1/H1/NSD2/EZH2 等多模型支持“confinement 与 Polycomb 3D contacts”这一原则。
读图要点:ED5A-B:其他 H3K27M lines 的 xenograft 与 cell-state 分析,支持 Fig.5 不是 BT245 单一模型。
读图要点:ED5C:patient scRNA 中 H3K27M 的 cPRC1-looping gene score 普遍较低。
读图要点:ED6:CBX-AM、CBX2/CBX4-KO、PHC2-L307R 的 controls。关键点是 PHC2-L307R 主要破坏 PHC2 聚集,而不是简单消除所有 cPRC1 binding。
读图要点:ED7:CBX2-KO 而非 CBX4-KO 显著延长 xenograft 生存,支持 CBX2 特异性。
创新点总结
| 创新点 | 为什么重要 | 对我有何用 |
|---|---|---|
| 把 H3K27me3 的“空间分布形态”作为核心变量 | 超越了传统“标记总量升降”的解释框架,提出 confinement/spreading 可调控 reader 的局部浓度。 | 做组学分析时不要只报 peak 数量或平均信号,可引入 peak breadth、domain spread、entropy/confinement 类指标。 |
| 连接 histone mark reader concentration 与 3D genome architecture | 说明表观修饰不只是线性调控基因表达,还可通过 reader 聚集组织核内 compartment/loop。 | 可迁移到 enhancer reader、DNA methylation reader 或 H3K9me3-HP1 体系,问“reader 是否被局部浓缩”。 |
| 用疾病突变作为机制工具 | H3K27M/EZHIP 虽是肿瘤驱动因素,但在本文中也被用作人为限制 H3K27me3 spreading 的自然实验。 | 疾病背景不只是样本来源,也可以被设计成扰动工具,用来构建近似因果对照。 |
| 多模型双向因果验证 | confined 模型、spreading 模型、cPRC1 直接扰动、体内 xenograft 互相咬合,证据链较完整。 | 自己的课题也可按“现象-机制候选-反向扰动-功能输出”搭证据链,而不是堆单一组学结果。 |
| 把 CBX2 与 PHC2 聚集性质拆开验证 | 不仅说明 cPRC1 binding 重要,还说明 oligomerization/aggregation 对 loop 维持有功能贡献。 | 在设计实验时优先找能拆分 binding、activity、aggregation 的 mutant 或 chemical probe。 |
可借鉴的研究思路
1. 从“总量”转向“分布熵/局部浓度”
很多组学变化不一定体现在全局 abundance,而体现在 peaks 是否从少数核心位点扩展到更宽区域。可在自己的项目中设计 confinement score、peak breadth、local concentration index 或 entropy-like metric。
2. 用 reader 的“有限资源分配”解释结构和功能
如果一个 reader 总量有限,binding sites 从窄变宽,就会出现 titration/dilution。这个思路可推广到 BRD4-acetylation、HP1-H3K9me3、MBD-DNA methylation 等体系。
3. 把多组学聚成“机制闭环”
本文每一步都用不同测量支持:ChIP/ChIC 定义 chromatin state,Hi-C 看 structure,PRO/RNA/scRNA 看表达和状态,xenograft 看体内功能。具体流程拆解见“生信与分析借鉴”。
4. 先做 unsupervised 分类,再回到特定机制
作者不是预设所有 H3K27me3 sites 都一样,而是用 chromatin features 做 UMAP/HDBSCAN,找出真正有 3D loop 的 cPRC1 subset。这个思路可作为后续多组学分层分析的模板。
5. 设计“拆机制”的 perturbation
PHC2-L307R 是漂亮的设计:尽量把 cPRC1 chromatin binding 和 SAM-domain oligomerization 分开,有助于证明聚集性质本身的作用。
生信与分析借鉴
A. 可迁移的分析问题
B. 本文生信流程拆解
| 分析层 | 输入数据 | 核心指标/方法 | 输出问题 |
|---|---|---|---|
| mark 分布 | H3K27me3 ChIP/ChIC | confinement score、peak breadth、domain spreading | 标记是集中在少数位点,还是铺展到邻近区域? |
| reader 占位 | CBX/PHC/RING1B profiles | CGI-level enrichment、overlap、cluster-specific signal | cPRC1 是被局部浓缩,还是整体 abundance 改变? |
| 3D interaction | Hi-C | aggregate peak analysis、pile-up、contact enrichment | 特定位点集合之间是否存在更强平均 contact? |
| 状态分类 | promoter/CGI 多组学特征矩阵 | UMAP/HDBSCAN 或类似无监督聚类 | 哪一类位点真正驱动 Polycomb loop? |
| 表达输出 | RNA-seq、PRO-seq、bulk/scRNA | gene set score、differential expression、cell-state projection | 结构变化是否对应发育分化程序被压制或释放? |
C. 读图时生信视角的必问三句
- 这个图是在比较总量、峰形、位点集合,还是三维 interaction?单位是什么?
- 位点集合是怎么定义的:所有 H3K27me3 sites,还是 cPRC1-rich subset?是否有选择偏倚?
- 效应是否在独立模型、反向扰动和功能输出中同向出现?
D. 若在自己的课题试做:最小可行清单
- 选一个 mark-reader 对,例如 H3K27me3-cPRC1、H3K27ac-BRD4 或 H3K9me3-HP1。
- 定义 disease/control 或 perturbation/control;先做 peak width、domain spread、reader peak concentration 的基础统计。
- 若有 Hi-C/PLAC-seq/HiChIP,围绕 reader-rich targets 做 pile-up;没有 3D 数据时,可先用公开数据或 promoter-enhancer contact 注释做探索。
- 把 target gene set 投射到 RNA-seq/scRNA-seq,看是否对应状态转换、分化程序或预后相关模块。
- 优先寻找一个能反向改变 mark spread 或 reader occupancy 的扰动模型,避免停留在描述性相关。
- 胸外科公共 bulk / scRNA 数据中,可先对 Polycomb / 发育分化 gene set 做 ssGSEA 方向筛查,借鉴 Fig.5h-j 的患者验证逻辑。
- 肺癌或 ESCC 队列里,可探索是否存在“抑制性 mark 更 confined + 3D hub 基因”的假说;这需要另行验证,不能直接套用 H3K27M 结论。
- 组内 HPC 可考虑固化一个最小 Snakemake 模块:从 CGI / peak set 到 cool 文件 pile-up,服务后续表观+3D 联合项目。
E. 与其他总结模块的分工
“可借鉴的研究思路”偏概念迁移;“方法速查”偏论文使用了哪些技术;本节则偏如何把这些技术转化为自己的分析流程和检查清单。
核心图模型复盘
把全篇压缩成一条因果链:H3K27me3 的分布形态不是背景现象,而是通过 reader complex 的局部浓度来调控三维基因组和细胞命运。
H3K27M、EZHIP、hiPS cell 中,H3K27me3 主要集中在 CGI/promoter 附近。
CBX2/4/8 与 PHC2 被集中招募,有限 reader pool 在少数位点形成高局部浓度。
CBX2 IDR 与 PHC SAM oligomerization 支持 cPRC1 foci 和远距离 contacts。
cPRC1-looping targets 多为发育/神经分化基因,H3K27M 中被更强压制。
扰动 CBX2、CBX-H3K27me3 binding 或 PHC2 aggregation,解除分化阻滞并削弱肿瘤形成。
| 证据节点 | 对应图 | 一句话结论 |
|---|---|---|
| 现象 | Fig.1 | H3K27me3 confinement 与 H3K27me3-CGI 之间更强 3D contact 相关。 |
| 机制候选 | Fig.2-3 | 真正增强 loop 的是 cPRC1-rich Polycomb target subset。 |
| 反向因果 | Fig.4 | 让 H3K27me3 spread 会稀释 cPRC1 并削弱 loop。 |
| 功能输出 | Fig.5-7 | cPRC1 loop 压住分化程序;打断 cPRC1 结构可促进分化、抑制 tumor phenotype。 |
技术路线与数据类型总结
这篇文章的强处不是某一个技术,而是把“表观修饰分布、reader 占位、三维结构、转录输出、体内表型”串成同一条证据链。
| 层级 | 主要数据/技术 | 本文回答的问题 | 读图重点 |
|---|---|---|---|
| Histone mark | H3K27me3 ChIP-seq/ChIC-seq、ChIP-Rx、MS normalization | H3K27me3 是 confined 还是 spreading?总量变化和分布变化能否分开? | 不要只看 peak 高低,要看 peak breadth、domain spread 和 confinement score。 |
| Reader occupancy | CBX2/4/8、PHC2、RING1B ChIP/ChIC/CUT&RUN;chromatin fraction MS | cPRC1 是总量变了,还是局部浓度变了? | 强调 MS 归一化后的 occupancy,和 H2AK119ub 的弱相关性。 |
| 3D genome | Hi-C matrix、aggregate peak/pile-up、FISH | Polycomb target 之间 contact 是否增强?是否是全局 TAD/compartment 改变? | 中心 enrichment 是平均 interaction;FISH 提供独立空间验证。 |
| Chromatin state | UMAP/HDBSCAN clustering of CGIs/promoters | 哪一类 Polycomb 位点真正形成 loop? | cPRC1 cluster 是关键,不是所有 H3K27me3 位点都等价。 |
| Expression/state | RNA-seq、PRO-seq、bulk/scRNA-seq、ssGSEA | loop targets 是否对应被压制的发育分化程序? | 从 nascent transcription 到 patient scRNA,观察 gene set 的一致方向。 |
| Perturbation/in vivo | CBX-AM、CBX2-KO、PHC2-L307R、SOX10 IF、xenograft survival/IHC | cPRC1 recruitment/aggregation 是否必要?是否影响肿瘤维持? | 区分 binding perturbation 与 aggregation perturbation,突出 CBX2 specificity。 |
开放问题与后续实验设想
下一步可做 multiplexed DNA FISH、live-cell tagged cPRC1 imaging、single-cell Hi-C 或 SPRITE/MERFISH 类空间验证,区分稳定结构、瞬时接触和群体平均。
可补 FRAP、1,6-hexanediol 敏感性、IDR mutant rescue、内源标签成像,并量化 condensate size、lifetime、fusion behavior。
可用 dCas9-EZH2/EED 或 locus-specific recruitment 在单个位点操控 spreading,观察 cPRC1 occupancy、local contact 和目标基因表达的时间顺序。
可做 CBX2/CBX4 domain-swap、IDR deletion、chromodomain mutant、rescue experiment,拆分 expression、binding specificity 和 aggregation capacity。
需要比较 H3K27M tumor cells、normal neural progenitors 和成熟 glial cells 对 CBX/cPRC1 perturbation 的敏感性,评估毒性和 lineage specificity。
可迁移到 HP1-H3K9me3、BRD4-acetylation、MeCP/MBD-DNA methylation,检验“mark spreading -> reader dilution -> 3D architecture”是否是通用机制。
不足、风险与开放问题
可能的不足
- Hi-C pile-up 是平均信号:中心 enrichment 强不等于每一对位点都形成稳定 loop,需要注意群体平均和细胞异质性。
- confinement score 与 pile-up 的因果方向仍需谨慎:两者同向变化不等于 mark 分布一定先于 3D contact;批次、细胞周期、细胞状态比例也可能影响 pile-up 强度。
- LLPS 证据主要是功能和分布推断:虽然 CBX2/PHC2 性质支持 condensate 模型,但严格的相分离物理验证在细胞内仍难完全证明。
- H3K27M 背景复杂:H3K27M 不只影响 H3K27me3 spreading,可能还有 PRC2 redistribution、developmental context、其他 chromatin state 的间接效应。
- 药物可转化性仍早:CBX-AM 是机制 probe,是否能成为治疗策略还需要药代、特异性、毒性和血脑屏障等验证。
讨论与延伸问题
- 如果 cPRC1 local concentration 是关键,是否能用 live-cell imaging 或 single-cell Hi-C 直接观察 loop/condensate 的异质性?
- H3K27me3 spreading 在不同细胞命运转换中是因还是果?是否存在主动调控 reader dilution 的发育机制?
- CBX2 为什么比 CBX4 更关键?是 expression、IDR/phase separation、靶点选择性,还是肿瘤 lineage-specific dependency?
- 把 H3K27me3 confinement 作为 biomarker,能否预测对 PRC1/CBX perturbation 的敏感性?